
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Summary
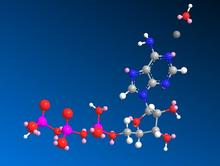
Quantum chemical calculations have proven to be a reliable tool for the prediction of thermodynamics quantities, particularly for smaller organic species. Computations of the thermodynamics of biomolecules in solution are rare in the literature. In this project, the equilibrium solvated structured of AMP, ADP, ATP, and associated molecules have been determined using density functional theory (DFT). In order to produce reliable estimates of the thermodynamics properties, careful attention is being paid to the convergence of the energy with respect to the basis set, and to the treatment of hindered rotors. When these concerns are fully addressed, it is expected that the results will compare favorably to the available experimental values.
Description
Goals:
- Demonstrate the validity of DFT methods for computing thermodynamics of biomolecules in solution.
- Identify new families of biomolecules for further study.
Research Activities:
- Seek improvements to the present methodology to improve results and reduce uncertainties.
Major Accomplishments
- A full set of structures has been optimized and initial estimates of thermochemistry have been made